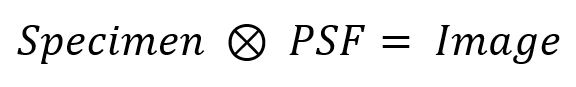Digital cameras have been the primary means for recording fluorescence microscope observations for several decades. The three main types of digital cameras used in scientific microscopy applications are CCD, EMCCD, and sCMOS cameras. Each one of these types of camera has its own advantages. The sCMOS cameras are capable of blazing fast frame rates (hundreds of frames per second) to capture millisecond-scale dynamics. EMCCD cameras have high quantum efficiency, large pixels, and low noise, making them the camera of choice when very high sensitivity is required such as for detecting single fluorescent molecules. For nearly everything else, the interline CCD camera is likely your best choice: providing a good balance between sensitivity and resolution and between relatively low noise and moderate speeds, the CCD-equipped microscope is the workhorse of many research labs.
Digital cameras have been the primary means for recording fluorescence microscope observations for several decades. The three main types of digital cameras used in scientific microscopy applications are CCD, EMCCD, and sCMOS cameras. Each one of these types of camera has its own advantages. The sCMOS cameras are capable of blazing fast frame rates (hundreds of frames per second) to capture millisecond-scale dynamics. EMCCD cameras have high quantum efficiency, large pixels, and low noise, making them the camera of choice when very high sensitivity is required such as for detecting single fluorescent molecules. For nearly everything else, the interline CCD camera is likely your best choice: providing a good balance between sensitivity and resolution and between relatively low noise and moderate speeds, the CCD-equipped microscope is the workhorse of many research labs.
Research-grade CCD cameras range in price from $2,000 to $20,000, depending on acquisition speed, read-out noise, and depth of cooling. Most high-performance CCD cameras are cooled to a temperature between 0 and -30˚C. Cooling adds the significant cost of a thermoelectric device and also requires that the sensor be hermetically sealed to prevent damaging condensation on the sensor. But how does the costly addition of cooling actually affect the performance of the CCD camera? For rigorous life sciences research, which applications require a cooled CCD, and which ones do not?
The Cameras
We compared an uncooled Lumenera Infinity 3-1UR (list $4250) against a cooled Roper Scientific Coolsnap HQ2 (list $16,840). The most relevant specifications for the two cameras are shown in Table 1. Both cameras incorporate the Sony ICX285 sensor, so some parameters such as the resolution, pixel size, and quantum efficiency (QE) will be identical. Other parameters such as the bit depth and read noise will be governed by the electronics used to digitize the signal. The biggest difference is in the dark current, which is reduced from about 1e- to 0.001e- by cooling the HQ2’s sensor.
| Specification |
Lumenera Infinity 3-1UR |
Roper Scientific Coolsnap HQ2 |
| Image Sensor |
Sony ICX285 |
Sony ICX285 |
| Resolution (pixels) |
1392 x 1040 |
1392 x 1040 |
| Pixel Size (um) |
6.45 x 6.45 |
6.45 x 6.45 |
| Quantum Efficiency (500nm) |
62% |
62% |
| Bit Depth |
14 bits |
14 bits |
| Full Well Capacity (e-) |
18,500 |
16,000 |
| Read Noise (e-) |
6 |
5.5 |
| Dark Current (e-/s) |
< 1 |
0.001 |
| Sensor Temperature |
+37˚C |
-30˚C |
Table 1: Comparison of camera specifications
Case Study #1: Fixed-cell microscopy

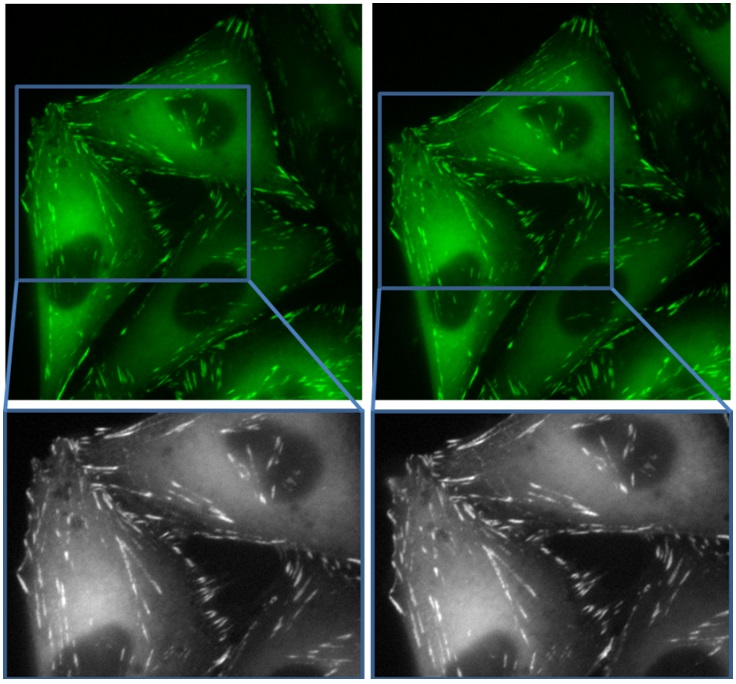
Figure 1: Alexa 488-labeled actin filaments in fixed endothelial cells imaged with A) cooled and B) uncooled cameras.
Fixed cells and tissues are labeled with relatively bright and stable fluorophores, making fluorescence microscopy on these samples a relatively routine endeavour. Figure 1 shows actin filaments in fixed endothelial cells labeled with Alexa Fluor 488 (FluoCells Prepared Slide #1, Life Technologies, F36924). The cells were imaged on a Zeiss AxioObserver inverted fluorescence microscope using an X-Cite 120 fluorescence lamp, a 40x/0.75NA Plan-Neofluar objective lens, and either the Infinity or Coolsnap camera (we consistently captured with the Coolsnap camera first). With the lamp set to 50% power, we used an exposure time of 1 second which filled most of the dynamic range of each camera without saturating. Under these imaging conditions, there was no discernible difference whatsoever between the images taken by the two cameras. Both cameras produced sharp, clear images of the actin filaments with no difference in noise or background level. We also reduced the exposure time incrementally from 1s down to 100ms to mimic imaging of dimmer fluorophores, but the two cameras produced identical results in all cases.
Case Study #2: Live-cell microscopy

Figure 2: Paxillin-GFP expressing CHOK1 cells imaged with a 3s exposure time on A) cooled and B) uncooled cameras.
Living cells with fluorescent protein tags are more difficult to image: the fluorescence is typically weaker than with fixed-cell probes, the illumination power is often lowered to minimize photobleaching and phototoxicity, and shorter exposures may be required to capture cellular dynamics. Figure 2 shows CHOK1 cells expressing the focal adhesion protein Paxillin-GFP (cells courtesy of Claire Brown, McGill University, Montréal, Canada). The cells were imaged on a Zeiss AxioObserver microscope using a 63x/1.4NA objective with the lamp power reduced to 25% and a 3 second exposure time. Again, both cameras were able to capture superb images of the focal adhesions under these conditions: there was no difference in the image quality despite the fact that one camera was cooled and the other was not.

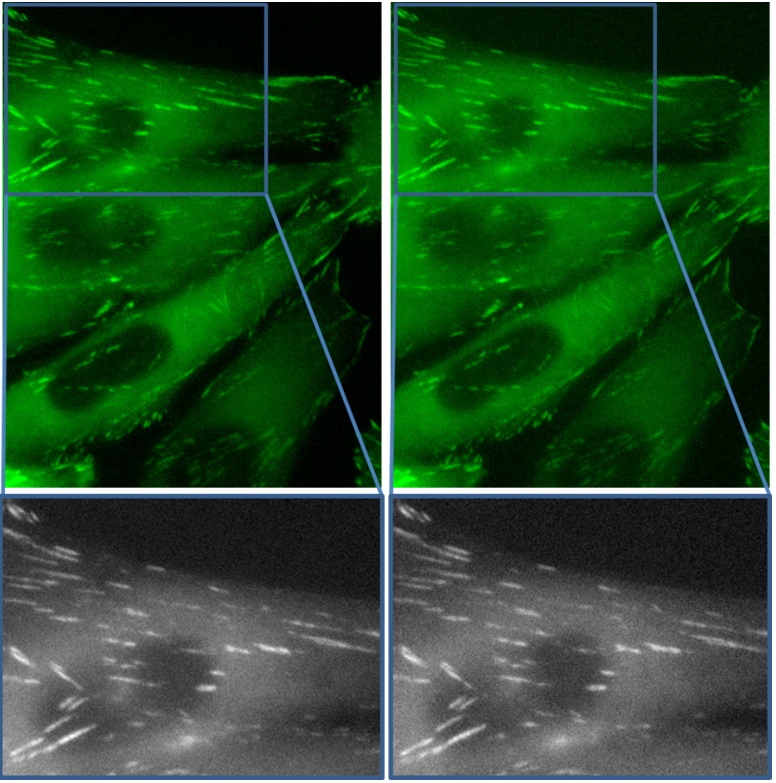
Figure 3: Paxillin-GFP expressing CHOK1 cells imaged with a 500ms exposure time on A) cooled and B) uncooled CCD cameras.
To achieve a faster frame-rate, we lowered the exposure time to 500ms and captured the low-light level images shown in Figure 3. Although obviously noisier, the two cameras are equally noisy and the focal adhesions are still readily trackable. The uncooled CCD camera achieves the same result as its considerably more expensive cooled counterpart for this challenging live-cell microscopy application.
Uncooled camera performance
Many microscopists assume that an uncooled CCD camera is not good enough for demanding live-cell imaging applications; but these experiments demonstrate that cooling the sensor is completely unnecessary. Why does the uncooled camera perform so well?
A good CCD camera for fluorescence microscopy should have a high Quantum Efficiency (a measure of the sensitivity of the sensor), high resolution (lots of pixels), high read-out speeds, and low noise. The first 3 parameters are completely independent of the camera temperature. Only the noise depends partly on temperature. CCD cameras have two main sources of noise:
- The read noise is the variation (or uncertainty) in reading out the values on the CCD sensor, primarily due to electronic components. Read noise varies with the read-out speed, but for these research-grade cameras it is generally limited to ± 6 electrons (e-) per pixel, independent of the exposure time. The maximum number of electrons that fit in each pixel of the image (called the full-well capacity) is more than 16,000 for the two cameras tested, so if you have enough light to fill at least half of the camera’s full-well capacity, the read noise is nearly negligible as in Figure 2. On the other hand, in Figure 3 where the signal is less than 10% of the full-well capacity, the read noise becomes apparent. Read noise, however, is still independent of the sensor temperature and is nearly identical for the two cameras tested.
- A dark current (or “dark charge”) accumulates in the sensor during longer exposures due to thermal processes, and the dark current noise is the statistical variation (or uncertainty) in the dark current level. The dark current noise does depend on the temperature of the sensor as well as the exposure time. You can measure and subtract the dark current background itself by closing the shutter and capturing a dark image, but the dark current noise has a randomness to it that can’t be subtracted. Cooling the camera can reduce the dark current level (and associated noise) to essentially zero for exposure times on the order of seconds. Even for the uncooled camera, however, the dark current noise of <1 is much lower than the read noise and therefore doesn’t contribute to the image.
In summary, cooling a CCD sensor only helps to lower the dark current and doesn’t significantly affect the other sensor parameters; but in the well-designed Infinity CCD, cooling wasn’t necessary to reduce the dark current well below the read noise of the camera, rendering the dark current noise negligibly low even for demanding live-cell fluorescence microscopy experiments.
Why do we still cool our CCD cameras?
Originally, CCD cameras had high dark current and it was necessary to cool them, particularly for astronomy applications that can require minute-long exposure times. But since about 1990, new technology for scientific-grade interline CCD sensors has lowered the dark current by about a factor of 1000 to well below the read noise of the camera. The low dark current makes cooling the sensor unnecessary even for rigorous fluorescence microscopy applications, provided exposure times are limited to no more than 10 seconds. It is likely that many users choose a cooled CCD camera even for routine fluorescence microscopy simply for historical reasons.
Cooling is still required in other disciplines, such as in vivo bioluminescence imaging or astronomy, where exposure times can be a full minute or longer. For very low-light level applications, such as single-molecule detection or fast spinning-disk confocal microscopy, the dominant noise component is still the read noise and not the dark current noise: cooling the sensor will not improve the Signal to Noise Ratio for these photon-starved applications. Instead, for low-light level applications, the interline CCD should be set aside in favour of the added sensitivity of an Electron-Multiplier CCD (EMCCD) camera. EMCCD sensors have such a low read noise that the dark current noise becomes dominant: cooling the EMCCD sensor to about -80˚C is indeed required to lower the dark current noise.
References
Pawley, J. B. (2006). More Than You Ever Really Wanted to Know About CCDs. Handbook of Biological Confocal Microscopy. J. B. Pawley. New York, Springer: 918-931.
Spring, K. R. (2000). “Scientific Imaging with Digital Cameras.” BioTechniques 29(1): 70-76.
Joubert, J., Y. Sabharwal and D. Sharma (2011). “Digital Camera Technologies for Scientific Bio-Imaging.Part 3: Noise and Signal-to-Noise Ratios.” Microscopy and Analysis Sept 2011.